
The Technology of Confocal Laser Scanning Microscopy (CLSM)
A point-scanning confocal system features multiple laser lines,1 galvanometric scanning mirrors2, beamsplitters, and dichroic elements3 to guide excitation and emission light on their respective paths.
A pinhole4 in the detection beam path blocks light from above or below the desired focal plane, producing an image of a thin optical section. By moving the focal plane up and down, researchers can obtain multiple virtual slices of a tissue—called a z-stack—which can be analyzed frame by frame or rendered in 3-D. The pinhole aperture size is configurable: by making the pinhole larger or smaller, spatial resolution and section thickness can be increased or decreased. But the microscope is still constrained by the diffraction limit of light; however small the pinhole, lateral resolution cannot surpass ~200 nm.
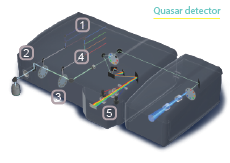
The instrument's detectors are also configurable. In the simplest optical configuration, emission light of the desired wavelength is directed through filters to one or more detectors. Live-cell and multicolor imaging applications require faster, more flexible detection systems capable of sampling much of the visual spectrum at once, such as the
34-channel Quasar detector diagrammed above.5
A Brief History of Confocal Microscopy
Marvin Minsky,
"Microscopy Apparatus"
U.S. Patent 3 013 467,
December 19, 1961.
Used with permission.
Given uniform excitation, as realized in widefield microscopy, all fluorescent molecules in a sample emit light simultaneously. Both in- and out-of-focus light emanating from these molecules reaches the detector, the latter causing blurring of the image. By contrast, confocal microscopy illuminates the sample point by point and blocks out-of-focus light using a pinhole positioned in focus with (confocal to) the focal plane, yielding a clearer picture.
Harvard University researcher Marvin Minsky first advanced this idea in 1957. His design imaged a sample point by point using an intense light source, assembling the data into a composite image.
In the following decades, multipoint (spinning disk) illumination strategies, developed in the 1960s, were succeeded by advances in laser excitation, new optical elements, and powerful computers. By 1987, the first confocal laser scanning microscope (CLSM)-a laser-based extension of Minsky's original idea-was commercially available.
Early designs held the light source steady and moved the sample on the stage. Today's CLSMs reverse that, using moving mirrors to scan the laser over the sample-a faster and more robust approach, especially for biological samples.
Platynereis larva.
Nathan Kenny, Kathryn McClelland, Sophie Miller, Marine Biological Laboratory, Woods Hole, MA, USA.
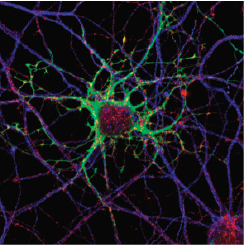
Primary neuronal cultures tained for Bornavirus antigens and tetanus toxin.
Caroline Charlier and Daniel Dunia, Inserm U1043, CPTP, Toulouse, France.
Going Live
The discovery and development of fluorescent proteins has greatly facilitated live-cell imaging and broadened the color palette for today's microscopists. The use of multiple markers has become standard, often with overlapping spectra.
Linear unmixing strategies can resolve overlapping colors by collecting a dataset representing a single optical section collected at different wavelengths, known as a lambda stack. By analyzing that stack pixel by pixel, the system can deconvolve the signal into distinct channels. Acquiring these multidimensional images over long time periods and large sample regions has driven the need for increased acquisition speed, as well as increased data-processing and data-handling capabilities.
Fluorescent proteins have broadened the color palette for today's microscopists.
Combining fluorescent molecules with novel imaging strategies reveals molecular dynamics. Photoactivatable fluorophores (which are dark until activated by a light pulse) and photoconvertible/photoswitchable fluorescent proteins (whose emission color can be altered with light) can highlight local sub-populations of molecules of interest in order to follow their dynamics. FRAP (fluorescence recovery after photobleaching) uses intense illumination to photobleach a defined sample region to measure how quickly new fluorophores migrate into or out of it, while FCS (fluorescence correlation spectroscopy) monitors molecules diffusing in and out of an illuminated region. RICS (raster image correlation spectroscopy) details the motion of molecules as they move across the scanned region (and between z frames) during image acquisition.
Confocal Microscopy for the 21st Century
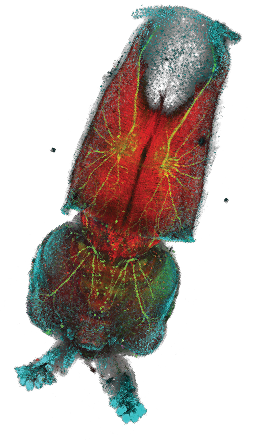
Loligo pealeiembryo.
Hooi Lynn Kee, Marine Biological Laboratory, Woods Hole, MA, USA.
Microscopy systems represent a balance between acquisition speed, resolution, and sensitivity, the so-called eternal triangle. Basically, acquiring an image faster means resolution and sensitivity inevitably suffer.
Alternative technologies enable researchers to "stretch" the corners of the eternal triangle, pictured below. Resonance scanning provides speed at the expense of collected signal. Superresolution strategies such as stimulated emission depletion microscopy (STED) circumvent the diffraction limit by effectively shrinking the fluorescent region using two overlapping lasers, sacrificing both imaging speed and sensitivity. Widefield superresolution techniques like PALM and STORM boost resolution at the expense of speed by localizing molecules using iterative imaging and bleaching strategies.


Download the full poster:
Download Full PosterOr select a section:
Download Poster Front Download Poster Introduction Download Poster AdvertisementsEvolution of fluorescent proteins and the fluorescent protein color palette
The discovery of fluorescent proteins has been a boon to CLSM. Green fluorescent protein (GFP), the first fluorescent protein discovered, and its multicolored relatives have allowed researchers to tag proteins and track their distribution and dynamics in vivo, a development that earned Roger Tsien, Martin Chalfie and Osamu Shimomura the 2008 Nobel Prize in Chemistry. Rather than affixing fluorophore-conjugated antibodies to a cell surface, or permeabilizing the membrane to direct such reagents towards intracellular targets, fluorescent proteins label proteins endogenously and in live cells. All that's required is to fuse the fluorescent protein coding sequence to a gene of interest.
Researchers use fluorescent proteins to monitor everything from gene expression to proteinprotein interactions to stem cell pluripotency. Initially, they were limited to just one color, green. Other proteins followed- variants in such basic colors as cyan (CFP), yellow (YFP), orange (OFP), and red (RFP). Today, the fluorescent protein palette is like a box of crayons, with such colorful proteins as mPium, mCherry, mBanana, and Emerald, the products of creative genetic manipulation.
Researchers have also boosted fluorescent protein brightness, and created such useful tools as photoactivatable and photoswitchable fluorescent molecules, such as PAmCherry and Dendra2, respectively; biochemical indicators such as GCaMP, a sensor of calcium signaling activity; and Arclight, a voltage indicator.
But fluorescent protein development has focused on more than spectral properties. Native fluorescent proteins are generally oligomers, but that can be problematic for many studies, especially those in which the proteins are expressed as fusions, so researchers have tweaked them to function as monomers- hence the "m" in proteins like mPJum. They also have built "split" protein isoforms that can be used to detect protein-protein interactions- an application called bimolecular fluorescence complementation
Linear Unmixing
Linear unmixing strategies use the known spectral properties of each fluorophore in a sample to back-calculate their individual contributions to each pixel of an image. The result is a dataset that resolves multiple seemingly indistinguishable signals into discrete color channels.
34-channel QUASAR
The Quasar detector spectrally separates emission light on a diffraction grating, then uses a series of sliders and prisms to direct specific spectral subsets to any of three possible detectors at once- two PMTs and a 32-channel high-sensitivity GaAsP detector array capable of high-speed, multicolor analysis.